- Matriz de sustitución
-
 Matriz PAM70 para 23 aminoácidos, calculada con el servicio web del Wageningen University Laboratory of Bioinformatics para tal fin.
Matriz PAM70 para 23 aminoácidos, calculada con el servicio web del Wageningen University Laboratory of Bioinformatics para tal fin.
En biología evolutiva una matriz de sustitución, o de puntuación, describe el ritmo al que un carácter en una secuencia cambia a otro carácter con el tiempo. Las matrices de sustitución se ven usualmente en el contexto de alineamiento de secuencias de aminoácidos o ADN, donde la similitud entre secuencias depende del tiempo desde su divergencia y de los ritmos de sustitución según se representan en la matriz.[1] Estas matrices se utilizan como parámetros de los algoritmos de alineamiento (por ejemplo los de Needlemann-Wunsch o Smith-Waterman), en los cuales cumplen el papel de asignar una determinada puntuación a cada emparejamiento entre los aminoácidos de las secuencias a alinear, contribuyendo así a la puntuación global del alineamiento.
Este tipo de matrices son más usuales en los alineamientos de secuencias de aminoácidos (proteínas) que en los de nucleótidos (ADN), ya que en este último caso suele utilizarse un sistema de puntuación mucho más simple para los emparejamientos entre los cuatro diferentes nucleótidos y que asigna, normalmente, una puntuación positiva para la coincidencia en el emparejamiento, una puntuación nula o negativa para la no coincidencia, y una puntuación negativa para los huecos o gaps.[2]
Contenido
Introducción
En el proceso de evolución, de una generación a la siguiente las secuencias de aminoácidos de las proteínas de un organismo son alteradas gradualmente a través de la acción de mutaciones de ADN. Por ejemplo, la secuencia
- ALEIRYLRD
podría mutar a la secuencia
- ALEINYLRD
en una generación, y posiblemente a
- AQEINYQRD
bajo un largo periodo de tiempo evolutivo. Al sustituir un determinado aminoácido por otro en una proteína, la probabilidad de ser el reemplazante no es la misma para todos los aminoácidos. Por ejemplo, en caso de mutación de un residuo hidrófobo como la valina es más probable que el nuevo residuo permanezca hidrófobo a que cambie, dado que su reemplazo por uno hidrófilo podría afectar al plegamiento o a la actividad de la proteína.[3]
Si nos encontramos con dos secuencias de aminoácidos, deberíamos ser capaces de investigar la posibilidad de que ambas se deriven de un ancestro común, u homólogo. Si podemos alinear las dos secuencias usando un algoritmo de alineamiento tal que las mutaciones requeridas para transformar una hipotética secuencia ancestro en ambas secuencias actuales pudieran ser evolutivamente plausibles, nos gustaría entonces asignar una alta puntuación a la comparación de las secuencias.
A este fin, construiremos una matriz cuadrada de, generalmente, 20x20 elementos (por los veinte aminoácidos usualmente contemplados, aunque nada impide contemplar los restantes y ampliar, en consecuencia, el orden de la matriz), donde la (i,j)-ésima entrada es igual a la probabilidad de que el i-ésimo aminoácido sea transformado al j-ésimo en una determinada cantidad de tiempo evolutivo (normalmente asumiremos que las sustituciones son simétricas: la probabilidad de sustitución de un aminoácido i por otro j será la misma que la probabilidad de sustitución del aminoácido j por el i, lo que nos resultará en matrices simétricas; no obstante, pueden contemplarse asimetrías si se toman en consideración las direcciones de cambio en un determinado árbol filogenético).[3] Hay diferentes maneras de construir tal matriz, que llamaremos matriz de sustitución. A continuación se exponen las más comunes.
Matriz identidad
La matriz de sustitución más simple posible sería una en la que cada aminoácido se considera máximamente similar a sí mismo, pero no es capaz de transformarse en cualquier otro aminoácido. La matriz aparecería como:

Esta matriz identidad tendrá éxito en el alineamiento de secuencias de aminoácidos muy similares, pero fracasará al alinear dos secuencias lejanamente relacionadas. Necesitamos contar con todas las probabilidades de una forma más rigurosa, y es cierto que, como veremos a continuación, un examen empírico de secuencias previamente alineadas trabaja mejor.
Matrices de log-probabilidades
Expresamos las probabilidades de transformación de un aminoácido en otro (es decir, cada uno de los elementos ai,j de la matriz de sustitución) mediante lo que se denomina puntuación por log-probabilidades. La matriz de puntuaciones se define entonces como

donde pi,j es la probabilidad, de acuerdo a las observaciones tomadas en consideración, de que el aminoácido i se transforme en el j, pi es la frecuencia de aparición del aminoácido i, y pj es la frecuencia de aparición del aminoácido j.[4]
De esta forma, el denominador pi·pj es la probabilidad de que ambos aminoácidos queden alineados por casualidad (matemáticamente, corresponde al producto de las probabilidades individuales de aparición); y el cociente entre ambas probabilidades puede resultar:
- Mayor que 1: la probabilidad observada de sustitución entre aminoácidos es superior a la aleatoria; según su magnitud, podría asumirse, en principio, que la evolución ha ido aceptando tal intercambio.
- Igual a 1: la sustitución entre uno y otro aminoácidos corresponde a la que puede encontrarse aleatoriamente a causa de mutaciones puntuales.
- Inferior a 1: la tasa de sustitución entre los aminoácidos es inferior a la que podría encontrarse aleatoriamente, por lo que según la magnitud del cociente podría asumirse, en principio, que esta sustitución no es aceptada evolutivamente de buen grado.
Aplicando de forma básica la teoría de la información, según la cual la cantidad de información H (p), en bits, que encontramos asociada a una probabilidad corresponde al logaritmo en base 2 de tal probabilidad (concretamente, H (p)=-log2p), tomamos el logaritmo del cociente para obtener, en definitiva, la similitud entre los aminoácidos en cuestión, representada por un número real que será positivo si el cociente visto es mayor que 1, negativo si es menor que 1, y nulo si el cociente es la unidad. La base del logaritmo no es especialmente importante, y puede verse con cierta frecuencia la misma matriz de sustitución expresada en bases logarítmicas diferentes, aunque computacionalmente puede interesarnos utilizar base 2 (para, como hemos visto, expresar la cantidad de información en bits) o base e (en cuyo caso la cantidad de información se mide en nats).[3]
Por la forma de calcular cada elemento, el conjunto de componentes de la matriz vendría expresado en números reales con un número indeterminado de decimales. Para su mejor tratamiento (tanto humano como informático), es aconsejable multiplicar cada elemento de la matriz por un factor de escala (lo que permite mantener la precisión) y redondear seguidamente al valor entero más próximo. El resultado es una matriz de enteros que retiene la precisión e información de los datos obtenidos originalmente. Sin embargo, y puesto que el factor de escala es arbitrario, encontraremos diferencias entre matrices calculadas desde una misma matriz inicial, por lo que una puntuación normalizada nos resultaría mucho más útil. Para esto último es necesario una constante específica para cada matriz, que se denomina lambda (λ), y que viene a resultar el equivalente al inverso del factor de escala.[3] [4]
Para calcular este valor λ, partimos de nuestra ecuación inicial, en la que incluimos el factor de escala como el inverso de λ y consideramos al logaritmo como de base e:

Tendremos, por lo tanto, que:

Como el logaritmo es de base e, y siendo n el número de aminoácidos contemplados (u orden de la matriz), aprovechando las propiedades de los logaritmos y utilizando (con los sumatorios: si la igualdad es cierta para todo i y j, lo será también para las sumas respectivas de ambos lados de la igualdad) todas las probabilidades implícitas en los elementos de la matriz, haremos:

Por definición de probabilidad, tenemos que la suma de todas las probabilidades
 , y por lo tanto:
, y por lo tanto:
Sustituyendo en esta última ecuación las "p" por las probabilidades conocidas, y las "a" por los resultados originalmente obtenidos, podremos resolverla para λ. Como es apreciable, cada matriz, con sus propias frecuencias o probabilidades para los aminoácidos, y sus propios resultados iniciales, tendrá una constante lambda diferente, pero que se aplicará como factor de escala a estos resultados iniciales de forma no arbitraria para conseguir la matriz de sustitución definitiva.
Principales matrices de sustitución
Las matrices de sustitución más utilizadas son las matrices PAM y las BLOSUM.[2] Ambas son matrices de log-probabilidades que se diferencian, principalmente, en los fundamentos para el cálculo inicial de las probabilidades de sustitución entre aminoácidos.
PAM
Una de las primeras matrices de sustitución, la PAM (Point accepted mutation, o mutación puntual aceptada), fue desarrollada por Margaret Dayhoff en los años 70 del pasado siglo. Esta matriz se calcula observando las diferencias en proteínas cercanamente relacionadas (con un mínimo del 85% de similitud).[4] La matriz PAM1 estima qué ritmo de sustitución debería esperarse si el 1% de los aminoácidos han cambiado, y se usa como base para el cálculo de otras matrices asumiendo que mutaciones repetidas seguirían el mismo patrón que las reflejadas en la matriz PAM1, así como que múltiples sustituciones pueden darse en el mismo sitio. Usando esta lógica, Dayhoff derivó matrices tan altas como PAM250, aunque normalmente se utilizan PAM30 y PAM70.
En resumen, Dayhoff realizó un trabajo con un fuerte componente teórico al asumir que se puede calcular una matriz para secuencias divergentes desde una matriz para secuencias cercanamente relacionadas elevando esta segunda matriz a una potencia.[4] Por ejemplo, podemos aproximar la matriz WIKI2 a partir de la WIKI1 expresando W2 = W12, donde W1 es WIKI1 y W2 es WIKI2. Así es, por ejemplo, como se calcula PAM250: elevando a la 250 potencia a PAM1.
Este modelo es el adecuado para seguir los orígenes evolutivos de las proteínas.[2]
BLOSUM
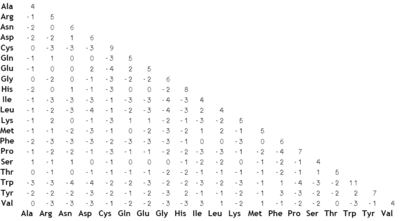 La matriz BLOSUM 62.
La matriz BLOSUM 62.La metodología de Dayhoff al comparar especies cercanamente relacionadas resultó no trabajar muy bien al alinear secuencias evolutivamente divergentes. Los cambios en las secuencias a lo largo de largas escalas de tiempo evolutivo no son bien aproximados combinando pequeños cambios que ocurren en cortas escalas de tiempo. La serie de matrices BLOSUM (de BLOck SUbstitution Matrix, o matriz de sustitución de bloques) corrige este problema.[4] Henikoff y Henikoff construyeron estas matrices usando múltiples alineamientos de proteínas evolutivamente divergentes. Las probabilidades usadas en los cálculos de la matriz se computan observando los "bloques" de secuencias conservadas encontrados en múltiples alineamientos de proteínas. Se asume que estas secuencias conservadas son de importancia funcional dentro de las proteínas relacionadas. Para reducir el sesgo por secuencias cercanamente relacionadas, los segmentos de un bloque con una identidad secuencial por encima de un determinado umbral fueron agrupados, ponderando con un factor de 1 a cada uno de tales grupos (Henikoff and Henikoff). Para la matriz BLOSUM 62, este umbral se fijó en el 62%. Se consideraron, entonces, pares de frecuencias entre los grupos, por lo que estos pares fueron sólo tomados en consideración entre segmentos con menos de un 62% de identidad. Se usarán matrices BLOSUM de numeración alta para alinear dos secuencias cercanamente relacionadas, mientras que se utilizarán números más bajos para secuencias más divergentes.
Se ha comprobado que la matriz BLOSUM 62 hace un excelente trabajo detectando similitudes en secuencias distantes, y esta es la matriz usada por defecto en las más recientes aplicaciones de alineamiento, como BLAST. En resumen, este modelo es adecuado para encontrar dominios conservados.[2]
Diferencias entre PAM y BLOSUM
- Las matrices PAM se basan en un modelo evolutivo explícito que asume que los intercambios entre aminoácidos ocurren bajo un proceso de Márkov que independiza los cambios en una determinada posición de los posibles cambios acaecidos previamente en ese mismo lugar, y que luego (puesto que se asume que estos cambios son de los primeros en ocurrir al considerar secuencias con un muy alto grado de similitud) se extrapolan para contemplar mayores distancias evolutivas, mientras que las matrices BLOSUM no se basan en ningún modelo explícito de evolución, y consideran secuencias de proteínas empíricamente relacionadas que comparten un antepasado común.[2]
- Las matrices PAM se basan en mutaciones observadas a través de un alineamiento global; esto incluye tanto regiones altamente conservadas como regiones mutables. Las matrices BLOSUM están basadas sólo en regiones altamente conservadas en series de alineamientos en los que no deben existir huecos (gaps).
- El método utilizado para considerar las sustituciones es diferente: al contrario que en la matriz PAM, el procedimiento de BLOSUM usa grupos de secuencias dentro de los cuales no todas las mutaciones ponderan igual.
- Números altos en el esquema de denominación de la matriz PAM denotan grandes distancias evolutivas, mientras que números altos en la matriz BLOSUM denotan alta similitud en las secuencias y, por lo tanto, distancias evolutivas más pequeñas. Por ejemplo: PAM150 se usa para mayores distancias que PAM100; BLOSUM 62 se usa para distancias más cortas que BLOSUM50.
Investigación actual
Las aproximaciones actuales más innovadoras incluyen la incorporación de la información de la estructura secundaria en las secuencias y en las matrices de sustitución, permitiendo la comparación de la secuencia problema con una biblioteca de plegamientos representativos.[5]
Véase también
- Algoritmo Needleman-Wunsch
- Algoritmo Smith-Waterman
- Alineamiento de secuencias
- Alineamiento múltiple de secuencias
- BLOSUM
- PAM
Referencias
- ↑ Attwood, T. K. (2002). «6». Introducción a la bioinformática. Prentice Hall. ISBN 84-205-3551-6.
- ↑ a b c d e Mount, D.W. (2004). «3 - Alignment of Pairs of Sequences». Bioinformatics. Sequence and Genome Analysis (2ª edición). CSHL Press. pp. 65. ISBN 0-87969-687-7.
- ↑ a b c d Korf, I et al. (2003). «4 - Sequence Similarity». En O'Reilly. BLAST. pp. 55. ISBN 0-596-00299-8.
- ↑ a b c d e Eddy, S.R. (2004). «Where did the BLOSUM62 alignment score matrix come from?». Nature Biotech. 22. 1035-6. http://informatics.umdnj.edu/bioinformatics/courses/5020/notes/BLOSUM62%20primer.pdf.
- ↑ Rice, D.W. y Eisenber, D. (1997). «A 3D-1D substitution matrix for protein fold recognition that includes predicted secondary structure of the sequence». Journal of Molecular Biology 267 (4). 1026-1038. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9135128&dopt=Abstract.
Bibliografía
- Altschul, S.F. Amino acid substitutions matrices from an information theoretic perspective. J. Mol. Biol. 219, 555-665 (1991).
- Dayhoff, M.O., Schwartz, R.M., Orcutt, B.C. A model of evolutionary change in proteins. En "Atlas of Protein Sequence and Structure" 5(3) M.O. Dayhoff (ed.), 345 - 352 (1978).
- Henikoff, S. and Henikoff, J. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA. 89(biochemistry): 10915 - 10919 (1992).
Enlaces externos


Wikimedia foundation. 2010.