- Enfermedad de Moyamoya
-
Enfermedad de Moyamoya
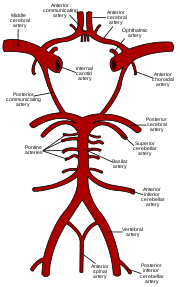 Representación esquemática del círculo de Willis, arterias cerebrales y contención de la sangre.
Representación esquemática del círculo de Willis, arterias cerebrales y contención de la sangre.
Contenido
Enfermedad de Moyamoya
La enfermedad de Moyamoya es llamada así debido a su apariencia angiográfica típica: Moyamoya, Japonés para “misty” (voluta de humo). Es una enfermedad que aun ofrece mas preguntas que respuestas sobre su etiología y tratamiento. Por razones desconocidas el paciente con Moyamoya sufre estenosis progresiva y oclusión de las arterias intracerebrales basales.La forma idiopática de la enfermedad fue reportada por primera vez en 1957 en Japón. Maki y Enomoto publicaron una revisión extensa en 1988. [2]
Epidemiología
Inicialmente fue descrita en Japón y se pensó que la enfermedad únicamente ocurría ahí, donde se considera que es más alta su incidencia [3] pero se encontró que también se presenta en caucásicos, Afro-americanos y nativos americanos. Al momento se han reportado casos en todo el mundo en muchos grupos étnicos, con mayor frecuencia se están detectando pacientes en la población americana y europea. [9] En Japón, es la enfermedad cerebro-vascular más frecuente en los niños, afecta dos veces más a las niñas que a los niños con una prevalencia aproximada de tres por cada 100,000. La prevalencia en Europa es de una décima parte de la identificada en Japón. En los niños Coreanos la enfermedad de Moyamoya es la enfermedad cerebrovascular más frecuente. [5] La prevalencia en California y Washington es de 0.086 por cada 100,000 habitantes. En este estudio la frecuencia fue de 4.6 para asiático-americanos, 2.2 para afro-americanos y 0.5 para hispanos. [1]
Se ha identificado una presentación bi-modal. El primer grupo son niños menores de 10 años que usualmente se presentan con manifestaciones isquémicas.
Causa
Aun cuando la causa de la enfermedad se desconoce se ha sugerido un fondo genético para el desarrollo de la enfermedad. Se han identificado loci por análisis de enlace en las formas familiares de enfermedad de Moyamoya, respaldando un patrón de herencia multifactorial. [5] Los hallazgos histo-patológicos sugieren la presencia de factores sistémicos que estimulan la proliferación de células de músculo liso y su migración de la capa media a la capa íntima para producir múltiples capas de fibras elásticas en la íntima. Las células de músculo liso producen metaloproteínasas de matriz (MMP)-2 y MMP-9, una deficiencia genética de cualquiera de las dos ocasiona disminución en la invasión de céluas de músculo liso in vitro, y la formación de hiperplasia de la íntima in vivo. Algunas enfermedades se presentan acompañando a la enfermedad de moyamoya como la neurofibromatosis tipo l, la anemia de células falciformes (drepanocitemia), y el síndrome de Down lo que sugiere un fondo congénito. Frecuentemente, una infección de vías respiratorias altas precede a las manifestaciones clínicas, generando la pregunta si algunos factores que incrementan la permeabilidad pueden causar una enfermedad adquirida o progresiva vascular.
La enfermedad expresa respuestas angio-ectásicas y neo-angiogénicas en la vasculatura pial y dural [7] Se ha identificado un sitio mayor para la dominancia autósomica de la enfermedad de Moyamoya en la región telomérica de 17q25 [6]
Manifestaciones clínicas
Cuatro tipos principales de manifestaciones clínicas:
- Síntomas debidos a HSA o hemorragia intracerebral (32.9 %)
- infarto cerebral (21.7 %)
- Ataques de Isquemia Cerebral Transitoria – AIT- (20.5 %)
- Convulsiones (11.9 %), o una combinación de estos síntomas (5.4 %).
- Los AIT, infartos, y convulsiones ocurren con mayor frecuencia en niños menores de 10 años; entre los 20 y los 40 años de edad, los síntomas son usualmente debidos a hemorragia cerebral. [1]
Historia Natural y Pronóstico
El pronóstico de la enfermedad de Moyamoya es difícil de predecir debido a que su historia natural no esta firmemente establecida. 50 a 66 % de los pacientes con Moyamoya que no son tratados sufrirán deterioro neurológico severo comparado con un 2.6 % de deterioro en un reporte de meta-análisis de un grupo de 1156 niños tratados quirúrgicamente [8]
La evolución depende de la presentación clínica: Los ataques de isquemia cerebral transitoria (AIT) y las crisis convulsivas sugieren una evolución relativamente benigna, con un 96% de los pacientes llevando una vida normal y un 37 % muestran un pobre desarrollo mental. [1]
El inicio de los síntomas antes de los seis años de vida, infartos cerebrales, y hallazgos angiográficos tipo lll tienen un pronóstico pobre, con trastornos severos mentales y en la función motora que ocurren en un 44 % de los pacientes. [1]
Diagnostico
El diagnostico se basa en las técnicas de neuroimagen como la angiografía convencional o la angio-resonancia, demostrándose la típica rete mirabille o micro-colateralización. Además micro-aneurismas asociados que pueden causar hemorragia se pueden detectar con estas técnicas.
En base a los hallazgos angiográficos, varios subgrupos y etapas se han definido: Tipo l consiste en únicamente estrechamiento en la mitad anterior del polígono de Willis (Bifurcación de la carótida, ACM proximal, y ACA). El tipo ll también incluye la arteria comunicante posterior (ACoP). Y, el tipo lll afecta también las arterias cerebrales posteriores (ACP). Como resultante, las arterias lentículo-estriadas están extremadamente dilatadas para brindar flujo colateral; pueden verse en la angiografía como los vasos Moyamoya que forman una red de micro-vasos. Pueden observarse también anastomosis trans-durales y leptomeníngeas.
Criterios Diagnosticos: [3]
A. La angiografía es esencial para el diagnostico y debe al menos presentar los siguientes hallazgos:
-
- 1. Estenosis u oclusión en la porción terminal de la arteria carótida y/o en la porción proximal de la arteria cerebral anterior y/o arteria cerebral media
-
- 2. Redes vasculares anormales adyacentes a la lesión estenótica u oclusiva en la fase arterial
-
- 3. Estos hallazgos deben estar presentes bilateralmente
B. Cuando la resonancia magnética y la Angio-Resonancia muestran los siguientes hallazgos, la angiografia no es obligatoria:
-
- 1. Estenosis u oclusión de la porción distal de la arteria carótida interna o de la arteria cerebral anterior y arteria cerebral media en la angio-resonancia
-
- 2. Red vascular colateral en los ganglios basales en la angio-resonancia. Note que una red vascular anormal puede diagnosticarse cuando más de dos ausencias de flujo en los ganglios basales de un lado son identificados en la resonancia magnética
-
- (1) y (2) se observan bilateralmente
C. Debido a que la etiología de esta enfermedad es desconocida, es necesario excluir las siguientes enfermedades:
-
- 1. Arterioesclerosis
-
- 2. Enfermedad Autoinmune
-
- 3. Meningitis
-
- 4. Tumor Cerebral
-
- 6. Enfermedad de Recklinghausen
-
- 7. Trauma Craneal
-
- 8. Radioterapia Craneal
-
- 9. Otras
Diagnóstico [4]
-
- 1. Caso definitivo: Reúne los criterios de (A) o (B) y (C). En los niños, sin embargo, un niño que reúne los criterios de (A) (1) y (2), o (B) (1) y (2) en un lado y con estenosis importante en la porción terminal de la arteria carótida interna en el lado opuesto, es también incluido.
-
- 2. Caso probable: Reúne los criterios de (A) (1) y (2) o (B) (1) y (2) y (C) (afectación unilateral).
Se pueden observar muchos patrónes de pseudo-moyamoya, en la infancia vasculopatía lentamente progresiva de los grandes vasos con colateralización. Sin embargo, la progresión paralela y simétrica bilateral, la angiogénesis transdural, la ausencia de circulación colateral leptomenínegea con gran contribución transdural, la apariencia normal de la vascularidad de la fosa posterior y el papel de los vasos lentículo-estriados sugiere una enfermedad de moyamoya verdadera [7]Diagnóstico Histo-Patológico
Los hallazgos histo-patológicos muestran engrosamiento de la intima, con tejido fibroso y depósito lipídico; la lamina elástica interna es tortuosa, y la media y adventicia son delgadas. Como resultante, la luz del vaso esta desplazada excéntricamente, y las arterias corticales muestran una capa intima edematosa.
Hallazgos Histopatológicos [3]
-
- Engrosamiento de la íntima con estenosis y oclusión resultante en la porción terminal de la arteria carotida interna, usualmente en forma bilateral. En ocasiones se identifican depósitos lipidicos en la intima en proliferación.
-
- Las arterias que constituyen el Poligono de Willis (ACA, ACM, y ACoP) muestran estenosis o grados variables de oclusión debido a engrosamiento fibro-celular de la íntima, una ondulación de la lamina elástica interna y adelgazamiento de la capa media
-
- Númerosos pequeños canales vasculares (perforantes y ramas anastomóticas colaterales) se observan alrededor del polígono de Willis
-
- Conglomerados reticulares de pequeños vasos se observan en la piamadre
Tratamiento
Medico
En ocasiones se dan anti-agregantes plaquetarios a los pacientes con Moyamoya, usualmente esta es la terapia elegida cuando la enfermedad es leve o el paciente tiene un riesgo quirúrgico elevado, pero existen pocos datos que demuestren su eficacia a corto o largo plazo. [9]
Los anticoagulantes como la warfarina son raramente utilizados en el tratamiento de niños con Moyamoya, debido a la dificultad para mantener niveles sanguíneos terapéuticos y el riesgo de hemorragia después de traumas leves. Se han utilizado heparinas de bajo peso molecular. Se han usado también antagonistas de los canales del calcio para intentar tratar la cefalea y prevenir la frecuencia y severidad de los episodios de isquemia cerebral transitoria refractaria.
Quirúrgico
Los procedimientos de revascularización quirúrgica son utilizados ampliamente en el tratamiento de la enfermedad de Moyamoya, particularmente en pacientes con deterioro cognoscitivo o con síntomas recurrentes y progresivos.
La cirugía de conexión anastomótica directa de la arteria temporal superficial a la arteria cerebral media, con frecuencia es técnicamente compleja de realizar en los niños debido al pequeño calibre de los vasos en la piel cabelluda y de la arteria cerebral media receptora.[9]
Los procedimientos de revascularización indirecta incluyen la encefaloduroarteriosinangiosis y la encefalomioarteriosinangiosis. Existen variantes de estos procedimientos, desde la simple realización de múltiples trepanos craneales sin sinangiosis de las arterias, la craneotomía con inversión de la dura con la esperanza de incrementar la neovascularidad del cerebro a partir de la dura. Algunos pacientes se estabililizan sin tratamiento, sin embargo, con frecuencia esto sucede después de que han desarrollado defectos neurológicos severos.[9]
Las complicaciones potenciales de la cirugía para el tratamiento de la enfermedad de Moyamoya incluyen: ACV isquémico, hematoma subdural espontáneo o traumático y hemorragia intra-cerebral,
Referencias
- Spetzler RF, Carter LP: Neurovascular Surgery
- Maki Y, Enomoto T: Moyamoya disease. Childs Nerv Syst 4:204, 1988
- Prevalence and Clinicoepidemiological Features of Moyamoya Disease in Japan Findings From a Nationwide Epidemiological Survey. Stroke. 2008;39:42-47
- Fukui M; Members of the Research Committee on Spontaneous Occlusion of the Circle of Willis (Moyamoya Disease) of the Ministry of Health and Welfare, Japan. Guidelines for the diagnosis and treatment of spontaneous occlusion of the circle of Willis (‘moyamoya’ disease). Clin Neurol Neurosurg. 1997;99(suppl 2): S238 –S240.
- Hyun-Seung Kang. Single Nucleotide Polymorphisms of Tissue Inhibitor of Metalloproteinase Genes in Familial Moyamoya Disease.Neurosurgery 58:1074-1080, 2006
- Mineharu Y. Autosomal dominant moyamoya disease maps to chromosome 17q25.3. Neurology 2008; 70: 2357– 2363
- Lasjaunias P, Berenstein A. Surgical Neuroangiography Vol. 3
- Fung LW, Thompson D, Ganesan V. Revascularisation surgery for paediatric moyamoya: a review of the literature. Childs Nerv Syst. 2005;21:358 –364.
- E. Steve Roach, MD, FAHA, Chair; Meredith R. Golomb, MD, MSc; Robert Adams, MD, MS, FAHA; Jose Biller, MD, FAHA; Stephen Daniels, MD, PhD, FAHA; Gabrielle deVeber, MD; Donna Ferriero, MD; Blaise V. Jones, MD; Fenella J. Kirkham, MB, MD; R. Michael Scott, MD, FAHA; Edward R. Smith, MD. Management of Stroke in Infants and Children A Scientific Statement From a Special Writing Group of the American Heart Association Stroke Council and the Council on Cardiovascular Disease in the Young. Stroke. 2008;39:2644-2691
Enlaces externos
http://www.emedicine.com/neuro/topic616.htm
http://iier.isciii.es/er/prg/er_bus2.asp?cod_enf=1804
http://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=ES&Expert=2573
Categoría: Enfermedades vasculares

Wikimedia foundation. 2010.