- Síndrome de Stevens-Johnson
-
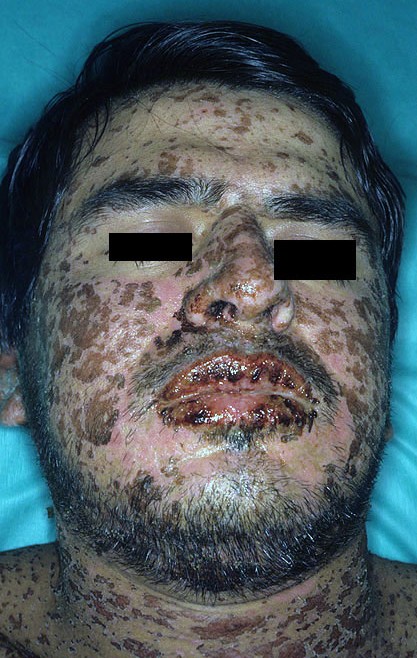
 El rostro de un hombre afectado por el síndrome de Stevens-Johnson.
El rostro de un hombre afectado por el síndrome de Stevens-Johnson.
El síndrome de Stevens-Johnson (SJS) y la necrólisis epidérmica tóxica (NET)[1] son dos formas de una enfermedad de la piel que amenaza a la vida, en la que la muerte celular hace que la epidermis se separe de la dermis. El síndrome se cree que es un complejo de hipersensibilidad que afecta a la piel y a las mucosas. Aunque la mayoría de casos sea idiopática (causa desconocida), la principal de las causas sabidas es la medicación, seguida de infecciones y, raras veces, cánceres.
Contenido
Clasificación
Existe un consenso en la literatura médica especializada que considera al síndrome de Stevens-Johnson como una forma más suave de necrólisis epidérmica tóxica (NET). Estas condiciones se reconocieron por primera vez en 1922.[2] Ambas enfermedades pueden confundirse con el eritema multiforme. El eritema multiforme a veces es causado por una reacción a una medicación, pero es más a menudo una reacción de hipersensibilidad tipo III a una infección (causado con frecuencia por el herpes simple) y es relativamente benigno. Aunque tanto SJS como NET también pueden ser causados por infecciones, son más a menudo los efectos adversos de medicaciones. Sus consecuencias son potencialmente más peligrosas que las de eritema multiforme.
Sintomatología
 Descamación de la mucosa de un afectado por el síndrome.
Descamación de la mucosa de un afectado por el síndrome. Conjuntivitis de una persona con el síndrome.
Conjuntivitis de una persona con el síndrome.El síndrome por lo general comienza con fiebre, dolor de garganta y fatiga, que es mal diagnosticado y por lo general se trataba con antibióticos. Las úlceras y otras lesiones empiezan a aparecer en las mucosas, casi siempre en la boca y labios, pero también en las regiones genitales y anales. Las que aparecen en la boca son por lo general sumamente dolorosas y reducen la capacidad del paciente de comer o beber. La conjuntivitis de los ojos ocurre en aproximadamente el 30% de los niños que desarrollan el SJS. Un brote de heridas redondas de aproximadamente una pulgada surge sobre la cara, el tronco, brazos y piernas, y las suelas de los pies, pero por lo general no en el cuero cabelludo.[3]
Patología
 Micrografía mostrando el grosor de la necrosis epidérmica con un estrato córneo con aspecto de tejido de esterilla y separación de la dermis y la epidermis. Biopsia de la piel. Tinción hematoxilina-eosina.
Micrografía mostrando el grosor de la necrosis epidérmica con un estrato córneo con aspecto de tejido de esterilla y separación de la dermis y la epidermis. Biopsia de la piel. Tinción hematoxilina-eosina.El síndrome, al igual que la necrólisis epidérmica tóxica y el eritema multiforme, está caracterizado por la confluente necrosis epidérmica con la inflamación mínima asociada. La agudeza es evidente observando el patrón de la capa córnea.
Causas
Se cree que el síndrome de Stevens-Johnson proviene de un desorden del sistema inmunológico.[3] La reacción inmune puede ser provocada por infecciones, drogas o medicaciones. En algunos grupos, la reacción a las drogas puede ser agravada por factores genéticos.
Infecciones
El síndrome de Stevens-Johnson puede ser causado por infecciones. Esto por lo general incluy infecciones comunes como el herpes simple, la gripe, las paperas, la enfermedad por arañazo de gato, la histoplasmosis, el virus de Epstein-Barr, mycoplasma pneumoniae o similares.
Medicamentos/drogas
Aunque el síndrome de Stevens-Johnson pueda ser causado por infecciones virales, malignidades o reacciones severas alérgicas a la medicación, la causa principal parece ser el empleo de medicinas de sulfas y antibióticos.
El SJS puede ser causado por la reacción adversa a medicamentos como alopurinol, fenitoína, ácido valproico, levofloxacino, diclofenaco, etravirina, isotretinoína, fluconazol,[4] valdecoxib, sitagliptina, oseltamavir, penicilinas, barbitúricos, sulfamida, azitromicina, oxcarbazepina, zonisamida, modafinilo,[5] lamotrigina, nevirapina, pirimetamina, ibuprofeno,[6] etosuximida, carbamazepina, nistatina y medicamentos contra la gota.[7] [8]
Entre los medicamentos que tradicionalmente se conocía que inducían al síndrome de Stevens-Johnson, eritema multiforme y la necrólisis epidérmica tóxica, se incluyen los sulfamidas, penicilinas, barbitúricos, lamotriginas y fenitoína. La combinación de lamotrigina con valproato de sodio aumenta el riesgo de padecer el síndrome.
Los antiinflamatorios no esteroideos son una causa rara del SJS en adultos; el riesgo es más alto para pacientes de mayor edad, mujeres y aquellos que están iniciando el tratamiento.[2] Típicamente, los síntomas del SJS inducidos por el consumo de drogas surgen una semana antes de comenzar la medicación. Las personas con lupus o con infecciones de VIH son más susceptibles al SJS inducido por drogas.[3]
El SJS también puede producirse por el consumo de cocaína.[9]
Genética
Algunas poblaciones estudiadas del oriente asiático (los Han y los tailandeses), el SJS inducido por carbamazepina y fenitoína está muy relacionado con HLA-B*1502 (HLA-B75), un serotipo HLA-B del serotipo más amplio HLA-B15.[10] [11] [12] Un estudio en Europa sugirió que el marcador génico solo era relevante para los asiáticos orientales.[13] [14]
Basándose en las conclusiones de Asia, estudios similares en Europa mostraron que un 61% de los pacientes con un SJS/TEN inducido por alopurinol presentaban el HLA-B58 (la frecuencia del fenotipo del alelo B*5801 en los europeos es normalmente del 3%). Un estudio concluyó: “ incluso cuando los alelos HLA-B actúan como fuertes factores de riesgo, en el caso del alopurinol, no son ni suficientes ni necesarios para explicar la enfermedad.”[15]
Tratamiento
El SJS constituye una emergencia dermatológica. Todas las medicaciones deberían ser interrumpidas, en particular aquellas que se sabe que causan reacciones del SJS. Los pacientes con infecciones documentadas del mycoplasma pueden ser tratados con macrólidos orales o doxiciclina oral.[3]
Al principio, el tratamiento es similar a esto para pacientes con quemaduras y el cuidado continuado solo puede ser de apoyo (por ejemplo terapia intravenosa y sonda nasogástrica o nutrición parenteral) y sintomático (por ejemplo el aclarado analgésico de boca para la úlcera bucal). Los dermatólogos y cirujanos tienden a discrepar sobre si la piel debería ser desbridada.[3]
Más allá de esta clase de cuidado de apoyo, no hay ningún tratamiento aceptado para el SJS. El tratamiento con corticosteroides es polémico. Recientes estudios retrospectivos sugirieron que los corticosteroides aumentan las permanencias en el hospital y los niveles de complicaciones. No hay ninguna prueba de corticosteroides para el SJS y puede ser manejado satisfactoriamente sin ellos.[3] Se han utilizado otros agentes, incluyendo la ciclofosfamida y la ciclosporina , pero ninguno ha demostrado demasiado éxito terapéutico. El tratamiento con inmunoglobulina intravenosa (IVIG) ha mostrado algún avance a la hora de reducir la longitud de la reacción y mejorar los síntomas. Otras medidas comunes de apoyo incluyen el empleo de anestésicos y antisépticos para el tratamiento del dolor, manteniendo un entorno caliente, y analgésicos intravenosos. Debería consultarse a un oftalmólogo inmediatamente, debido a que el SJS con frecuencia causa la formación de tejido cicatrizado dentro de los párpados, llevando a una vascularización de la córnea, visión perjudicada y una multitud de otros problemas oculares.
Prognosis
El SJS propiamente dicho (con menos del 10% de la superficie de cuerpo implicada) tiene un índice de mortalidad de alrededor del 5 %. El riesgo de muerte se estima usando la escala de SCORTEN, que toma un número de indicadores de pronóstico a tener en consideración.[9] Otras consecuencias pueden ser el fallo o daño de órganos, el rasgado de córnea y la ceguera.
Epidemiología
El síndrome de Stevens-Johnson es de condición rara, con una incidencia relatada de alrededor de 2,6[3] a 6,1[2] casos por millón de personas por año. En los Estados Unidos, hay aproximadamente 300 nuevos diagnósticos por año. La condición es más común en adultos que en niños. Las mujeres se ven afectadas más a menudo que los hombres, con casos que ocurren en una proporción de dos a uno (2:1).[2]
Historia
El síndrome de Stevens-Johnson recibe su nombre por Albert Mason Stevens y Frank Chambliss Johnson, pediatras estadounidenses que en 1922 publicaron conjuntamente una descripción del desorden en el American Journal of Diseases.[16] [17] [18]
Casos notables
- Padma Lakshmi actriz, modelo, celebridad y escritora de libros de cocina;[19]
- Manute Bol, jugador profesional de baloncesto y miembro de los equipos de la NBA Washington Bullets, Golden State Warriors, Philadelphia 76ers y Miami Heat, quien murió de complicaciones.[20]
Referencias
- ↑ Merck Manual: Stevens-Johnson syndrome
- ↑ a b c d «Severe adverse skin reactions to nonsteroidal antiinflammatory drugs: A review of the literature». American Journal of Health-System Pharmacy 67 (3): pp. 206–213. 2010. doi:. PMID 20101062.
- ↑ a b c d e f g Tigchelaar H, Kannikeswaran N and Kamat D (1 de diciembre de 2008). «Stevens-Johnson Syndrome: An Intriguing Diagnosis». Consultant for Pediatricians. http://www.consultantlive.com/consultant-for-pediatricians/article/1145470/1403936.
- ↑ Medsafe Data Sheet March 8, 2005. Accessed April 26, 2007.
- ↑ US FDA 2007 Safety Alerts for Drugs, Biologics, Medical Devices, and Dietary Supplements
- ↑ Raksha MP, Marfatia YS (2008). «Clinical study of cutaneous drug eruptions in 200 patients». Indian J Dermatol Venereol Leprol 74 (1): pp. 80. doi:. PMID 18193504.
- ↑ Fagot J, Mockenhaupt M, Bouwes-Bavinck J, Naldi L, Viboud C, Roujeau J (2001). «Nevirapine and the risk of Stevens–Johnson syndrome or toxic epidermal necrolysis». AIDS 15 (14): pp. 1843–8. doi:. PMID 11579247.
- ↑ Devi K, George S, Criton S, Suja V, Sridevi P (1 de septiembre de 2005). «Carbamazepine--the commonest cause of toxic epidermal necrolysis and Stevens–Johnson syndrome: a study of 7 years». Indian J Dermatol Venereol Leprol 71 (5): pp. 325–8. doi:. PMID 16394456. http://www.ijdvl.com/article.asp?issn=0378-6323;año=2005;volumen=71;número=5;spage=325;epage=328;aulast=Devi.
- ↑ a b Stevens–Johnson Syndrome - http://emedicine.medscape.com/article/1197450-overview
- ↑ Chung WH, Hung SI, Hong HS, et al. (April 2004). «Medical genetics: a marker for Stevens–Johnson syndrome». Nature 428 (6982): pp. 486. doi:. PMID 15057820.
- ↑ Locharernkul C, Loplumlert J, Limotai C, et al. (July 2008). «Carbamazepine and phenytoin induced Stevens–Johnson syndrome is associated with HLA-B*1502 allele in Thai population». Epilepsia 49 (12): pp. 2087. doi:. PMID 18637831.
- ↑ Man CB, Kwan P, Baum L, et al. (May 2007). «Association between HLA-B*1502 allele and antiepileptic drug-induced cutaneous reactions in Han Chinese». Epilepsia 48 (5): pp. 1015–8. doi:. PMID 17509004.
- ↑ Alfirevic A, Jorgensen AL, Williamson PR, Chadwick DW, Park BK, Pirmohamed M (September 2006). «HLA-B locus in Caucasian patients with carbamazepine hypersensitivity». Pharmacogenomics 7 (6): pp. 813–8. doi:. PMID 16981842.
- ↑ Lonjou C, Thomas L, Borot N, et al. (2006). «A marker for Stevens–Johnson syndrome ...: ethnicity matters». Pharmacogenomics J. 6 (4): pp. 265–8. doi:. PMID 16415921.
- ↑ Lonjou C, Borot N, Sekula P, et al. (February 2008). «A European study of HLA-B in Stevens–Johnson syndrome and toxic epidermal necrolysis related to five high-risk drugs». Pharmacogenet. Genomics 18 (2): pp. 99–107. doi:. PMID 18192896.
- ↑ Stevens AM, Johnson FC (1922). «A new eruptive fever associated with stomatitis and ophthalmia; report of two cases in children». Am J Dis Child 24: pp. 526–533.
- ↑ Stevens–Johnson syndrome - Definitions from Dictionary.com
- ↑ American journal of diseases of children. American Medical Association. 1922. pp. 526–. http://books.google.com/books?id=E90fAAAAIAAJ&pg=PA526. Consultado el 5 June 2010.
- ↑ Jess Cartner-Morley, "Beautiful and Damned", The Guardian, 8 de abril de 2006
- ↑ FanHouse Staff (19). «Manute Bol Dies at Age 47». Fanhouse. Consultado el 20 de junio de 2010.
Enlaces externos
 Wikimedia Commons alberga contenido multimedia sobre Síndrome de Stevens-Johnson. Commons
Wikimedia Commons alberga contenido multimedia sobre Síndrome de Stevens-Johnson. Commons
Categorías:- Enfermedades cutáneas
- Síndromes
- Enfermedades infecciosas



Wikimedia foundation. 2010.