- Síndrome de Miller-Dieker
-
Síndrome de Miller-Dieker 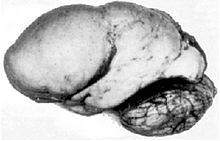
Cerebro con lisencefalia.Clasificación y recursos externos CIE-9 758.33 OMIM 247200 DiseasesDB 29494 MeSH D054221 Sinónimos Síndrome de lisencefalia de Miller-Dieker;
Síndrome de deleción hemicigótica 17p13,3 Aviso médico
Aviso médico El síndrome de Miller-Dieker, en ocasiones también denominado síndrome de lisencefalia de Miller-Dieker o síndrome de deleción hemicigótica 17p13,3,es una rara enfermedad congénita y hereditaria. Los afectados presentan problemas en el desarrollo del sistema nervioso central, lo que conduce a alteraciones severas en la función neurológica. Se caracteriza por presentar una lisencefalia tipo I, asociada a malformaciones craneofaciales como hundimiento bitemporal y mandíbula pequeña.
Contenido
Historia y epidemiología
En el año 1963, fue descrito por primera vez por J. Q. Miller.[1] Más tarde, H. Dieker y alumnos describieron el mismo conjunto de alteraciones, entre los que se incluía la lisencefalia.[2] K.L Jones completó el cuadro clínico, denominándolo como síndrome de Miller-Dieker para diferenciarlo de otras patologías que cursan con lisencefalia.[3]
Los estudios epidemiológicos arrojan una prevalencia global estimada de 0,3 casos por cada 100.000 nacidos.[4]
Etiopatogenia
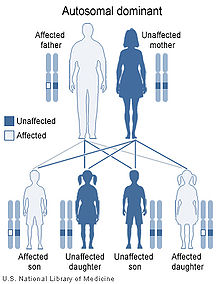 Patrón de herencia autosómica dominante.
Patrón de herencia autosómica dominante.
Este síndrome está causado por una mutación en el brazo corto del cromosoma 17. El gen afectado se denomina LIS1 (17p13,3) y codifica la proteína PAFAH1B1. Se han descrito diferentes mutaciones en los afectados por esta patología, tanto espontáneas como hereditarias. Entre las mutaciones de novo se encuentran el cromosoma 17 en anillo y las deleciones de la región terminal de este cromosoma. Las mutaciones hereditarias, siguen un patrón de herencia autosómico dominante, es decir, son necesarias ambas copias del gen para que sea funcional. Con una sola copia del gen afectado, se padece la enfermedad, lo que se denomina haploinsuficiencia. El portador se trata de una persona que no muestra síntomas debido a que su mutación es una translocación recíproca equilibrada con otro cromosoma. Este portador tendría entonces un cromosoma con ningún gen y otro con dos (está compensada). Otro tipo de portador es aquel que posee un cromosoma 17 recombinante, en el que la localización del gen no es la misma en ambos pares de cromosomas, produciendo durante la meiosis gametos con la falta de este gen.[3]
Sin embargo, el gen LIS1 no causa por sí mismo el resto de manifestaciones del cuadro clínico. Mutaciones aisladas en el mencionado gen están presentes en casos de lisencefalia tipo 1. Las demás anomalías pueden ser explicadas debido a la afectación de los genes contiguos al gen LIS1. En la región 17p13.3 en la que se produce la deleción, han sido identificados otros 50 genes.
La lisencefalia se produce debido a un fallo durante la migración neuroblástica durante la formación del sistema nervioso. El proceso de migración comienza, a las cuatro semanas de gestación y acaba alrededor del primer año de vida, de tal forma que casi ninguna neurona del cerebro de un adulto se encuentra en el mismo sitio donde comenzó a desarrollarse.
El factor activador de plaquetas acetilhidroxilasa (PAFAH), es una enzima que hidroliza al factor activador de plaquetas, que es un inhibidor de la migración neuroblástica. Además el PAFAH se encuentra asociado a los microtúbulos interviniendo en su estabilización y reorganización durante el desplazamiento neuronal. El gen faltante en el síndrome de Miller-Dieker codifica la isoforma presente en el cerebro de la subunidad β del PAFAH.[5]
Cuadro clínico
La principal manifestación del síndrome en el sistema nervioso central es la lisencefalia, generalmente severa, de tipo I, con frecuencia asociada a una falta total o parcial del cuerpo calloso (agenesia), adelgazamiento de la corteza cerebral, cisura de Silvio abierta y pequeñas calcificaciones en la línea media del tercer ventrículo.[6] La lisencefalia se caracteriza por la ausencia de circunvoluciones en la corteza cerebral, suele afectar a todo el encéfalo, incluido el cerebelo; presenta diferentes grados de afección, que van desde agiria (ausencia total de las circunvoluciones cerebrales), hasta paquigiria (pocos surcos con circunvoluciones de gran tamaño). Produce retraso mental, parálisis cerebral infantil, microcefalia, convulsiones, falta de regulación de la temperatura (sin homeostasis), rechazo al alimento, apneas y suele reducir la esperanza de vida. La lisencefalia tipo I es aquella en la que se pueden distinguir cuatro capas celulares de la corteza cerebral en lugar de las seis normales y presenta una escasa afectación del cerebro.[3]
Debido a la grave desestructuración cerebral aparece un retraso grave en la adquisición de las habilidades que requieren la coordinación de la actividad muscular y mental (psicomotor) y un retraso mental severo, además de tetraparesia (parálisis incompleta o ligera de los cuatro miembros) flácida y epilepsia rebelde al tratamiento que suele debutar como un síndrome de West.[6] Algunos pacientes solo consiguen desarrollar la habilidad para sonreir, mantener la vista fija y dar respuestas motoras inespecíficas.[5]
Externamente se pueden observar rasgos cráneo-faciales dismórficos: frente prominente y estrecha, estrechamiento o hundimiento bitemporal, surco medial en la frente, nariz pequeña con aberturas nasales antevertidas, labio superior prominente con filtro largo, micrognatia (mandíbula anormalmente pequeña), orejas de implantación baja y erupción de la dentición decidua retrasada.[3]
Otros signos que puede presentar un afectado por este síndrome son:
- retraso del crecimiento;
- polihidramnios (aumento anormal del líquido en la cavidad ammiótica) y disminución de los movimientos fetales;
- paladar ojival: en forma de bóveda;
- onfalocele: defecto de línea media en la pared abdominal anterior del abdomen, a nivel del ombligo;
- cardiopatías congénitas: ductus arterioso persistente, defectos del septo ventricular;
- anomalías urogenitales donde se destaca la criptorquidia (la más frecuente) agenesia renal e hidronefrosis;
- anomalías digitales: polidactilia, clinodactilia (arqueamiento permanente de un dedo), camptodactilia (flexión permanente de uno o más dedos) y dermatoglifos anormales;
- anomalías oculares: defectos del iris, telecantus (anomalía del párpado que consiste en una deformidad que aumenta la distancia del ángulo interno del ojo a la nariz), hipertelorismo, blefaroptosis y alteraciones de la vascularización retiniana.
Diagnóstico
El diagnóstico requiere realizar estudios de diagnóstico por imagen. A través de un escáner cerebral y, sobre todo, de una resonancia nuclear magnética sirven para confirmar las malformaciones cerebrales asociadas. Estos hallazgos junto a los rasgos faciales dismórficos son criterios necesarios y suficientes para hacer un diagnóstico. Se ha descrito un patrón electroencefalográfico característico que puede ayudar al diagnóstico diferencial: EEG con ritmos muy rápidos de entre 10 y 15 Hz y de voltaje muy alto y sin variación durante el sueño.[7] Para confirmar el diagnóstico se hace imprescindible un examen citogenético que desvele la característica microdeleción que se da en cromosoma 17 en los afectados por este síndrome.[5]
Es posible un diagnóstico prenatal. En caso de riesgo de ser portador, se puede realizar una biopsia o amniocentesis, realizando un análisis citogenético de las muestras. Este síndrome también puede detectarse mediante ecografía, pero es a partir del segundo trimestre cuando se sospecha lisencefalia al no detectarse los surcos de la corteza cerebral. Antes de las 20 semanas no son evidentes las circunvoluciones. Además, el retraso en el crecimiento fetal y polihidramnios durante el tercer trimestre es un posible signo de la enfermedad.[8] [9]
Tratamiento y pronóstico
No existe tratamiento curativo de la enfermedad, siendo el tratamiento puramente sintomático. La mayoría de los afectados precisan de medicamentos para controlar las convulsiones. Además suele ser necesario la alimentación mediante una sonda nasogástrica.[5] El síndrome de Miller-Dieker tiene mal pronóstico y los pacientes fallecen normalmente durante los dos primeros años de vida.[8]
Referencias
- ↑ Miller, JQ (1963). «Lissencephaly in 2 sibligs». Neurology. PMID 14066999.
- ↑ Dieker, H., Edwards, R. H., ZuRhein, G., Chou, S. M., Hartman, H. A., Opitz, J. M. (1969). «The lissencephaly syndrome». New York: National Foundation-March of Dimes II: pp. 53-64.
- ↑ a b c d Lyons Jones, Kenneth (2007). «Hallazgos cerebrales y/o neuromusculares infrecuentes con defectos asociados» (en español). Smith Características Reconocibles de La Malformacion Humana (6ª edición). Elsevier. pp. 208. ISBN 84-8174-947-8. http://books.google.es/books?id=wWVVPThnScEC&pg=PA207&dq=S%C3%ADndrome+de+Miller-Dieker&hl=es&ei=e3o7TfyvIYj_4AaTjczgCg&sa=X&oi=book_result&ct=result&resnum=1&ved=0CCsQ6AEwAA#v=onepage&q=S%C3%ADndrome%20de%20Miller-Dieker&f=false. Consultado el 24 de enero de 2011.
- ↑ Informes periódicos de Orphanet, Serie de Enfermedades raras (Noviembre de 2010). «Prevalence of rare diseases: Bibliografic data». Consultado el 24 de enero de 2011.
- ↑ a b c d Margaret W. Thompson, Robert L. Nussbaum, James Scott Thompson, Roderick R. McInnes, Huntington F. Williard (2008). «27. Síndrome de Miller-Dieker» (en español). Genética en medicina (7ª edición). Elsevier. pp. 286. ISBN 978-84-458-1225-9. http://books.google.es/books?id=gjklctElEywC&pg=PA286&dq=S%C3%ADndrome+de+Miller-Dieker&hl=es&ei=e3o7TfyvIYj_4AaTjczgCg&sa=X&oi=book_result&ct=result&resnum=2&ved=0CDAQ6AEwAQ#v=onepage&q=S%C3%ADndrome%20de%20Miller-Dieker&f=false. Consultado el 24 de enero de 2011.
- ↑ a b Romina Romero, María Corfio (2007). «Síndrome de Miller Dieker». Anacem I. http://revista.anacem.cl/AC%203.pdf.
- ↑ Villarejo Ortega, Francisco (1988) (en español). Tratamiento de la epilepsia (1ª edición). Díaz de Santos. pp. 144. ISBN 84-7978-325-7. http://books.google.es/books?id=253QHewpg_QC&pg=PA144&dq=S%C3%ADndrome+de+Miller-Dieker&hl=es&ei=j0E8Tb3XI9SG5Abxz736Cg&sa=X&oi=book_result&ct=result&resnum=3&ved=0CDIQ6AEwAjgU#v=onepage&q=S%C3%ADndrome%20de%20Miller-Dieker&f=false. Consultado el 24 de enero de 2011.
- ↑ a b Callen, Paul W (2009). «Síndromes fetales» (en español). Ecografía en obstetricia y ginecología (5ª edición). Elsevier. pp. 131. ISBN 978-84-458-1934-0. http://books.google.es/books?id=bQXWSrxi40AC&pg=PA131&dq=sindrome+de+miller-dieker&hl=es&ei=oqA9TbnfEpOs8QP4sIzVCA&sa=X&oi=book_result&ct=result&resnum=3&sqi=2&ved=0CDkQ6AEwAg#v=onepage&q&f=false. Consultado el 24 de enero de 2011.
- ↑ Klatt, EC (2007) (en español). Atlas de anatomía patológica (1ª edición). Elsevier. pp. 454. ISBN 978-84-8086-275-2. http://books.google.es/books?id=Z84eZf6V6xsC&pg=PA454&dq=S%C3%ADndrome+de+Miller-Dieker&hl=es&ei=j0E8Tb3XI9SG5Abxz736Cg&sa=X&oi=book_result&ct=result&resnum=8&ved=0CEwQ6AEwBzgU#v=onepage&q=S%C3%ADndrome%20de%20Miller-Dieker&f=false. Consultado el 24 de enero de 2011.
Categorías:- Síndromes
- Enfermedades genéticas
- Enfermedades hereditarias
- Enfermedades neurológicas
- Enfermedades raras
- Enfermedades epónimas
- Enfermedades congénitas


Wikimedia foundation. 2010.